 Получите свидетельство
Получите свидетельство Вход
Вход

ГОСУДАРСТВЕННОЕ БЮДЖЕТНОЕ ПРОФЕССИОНАЛЬНОЕ ОБРАЗОВАТЕЛЬНОЕ УЧРЕЖДЕНИЕ КАМЧАТСКОГО КРАЯ
«КАМЧАТСКИЙ МЕДИЦИНСКИЙ КОЛЛЕДЖ»
МЕТОДИЧЕСКАЯ РАЗРАБОТКА
ПРАКТИЧЕСКОГО ЗАНЯТИЯ
ОП. 04 Генетика с основами медицинской генетики
по теме: «Генные болезни»
для специальности: код 34.02.01 специальность Сестринское дело
Уровень подготовки: базовый
|
Рассмотрена на заседании цикловой комиссии общепрофессиональных дисциплин Протокол № ____ «__» ________2026 г. Председатель ЦМК_______/Г.В. Яковишин |
УТВЕРЖДАЮ Заместитель директора по УМР __________/Ю.И. Климова «___» ______ 2026 г. |
Составители:
Левенец О.В. – преподаватель высшей квалификационной категории ГБПОУ КК «Камчатский медицинский колледж»
г. Петропавловск-Камчатский, 2026
Содержание
1. Пояснительная записка………………………………………………………..3
2. Структурно-логическая схема занятия………………………………………8
3. Список использованных источников……………………………………….10
4. Приложение А. Контрольные вопросы для фронтального опроса………12
5. Приложение Б. Задание по определению фенотипов……………………...16
6. Приложение В. Таблица……………………………………………………..26
7. Приложение Г. Ситуационные задачи……………………………………....28
8. Приложение Д. Физкультминутка…………………………………………...33
9. Приложение Е. Текущий срез знаний……………………………………….34
10. Приложение Ж. Рефлексия…………………………………………………40
11. Приложение З. Оценочный лист……………………………………………41
12. Приложение И. Внеаудиторная самостоятельная работа…………………42
ПОЯСНИТЕЛЬНАЯ ЗАПИСКА
Методическая разработка практического занятия по теме: «Генные болезни» разработана в соответствии с ФГОС СПО специальности 34.02.01 «Сестринское дело» и рассчитана на одно практическое занятие.
Данная тема входит в изучение ОП.04. Генетика с основами медицинской генетики.
Методическая разработка практического занятия рассчитана на 2 академических часов в соответствии с календарно-тематическим планом программы учебной дисциплины.
Актуальность темы:
Моногенные синдромы и болезни (МБ), или генные
(так их называют за рубежом), заболевания, подчиняются
менделевскому наследованию, в их основе лежат единичные генные или точковые мутации. МБ составляют значительную долю наследственной патологии и насчитывают сегодня более 4500 заболеваний. По данным литературы, в разных странах они выявляются у 30–65 детей в расчете
на 1000 новорожденных, что составляет 3,0–6,5%, а в
структуре общей смертности детей до 5 лет на их долю
приходится 10–14%. Многие МБ, несмотря на достаточно
высокий уровень медико-биологических знаний, представляют значительные трудности в своевременной диагностике и эффективном лечении и часто приводят к значительному нарушению качества жизни больных, инвалидизации и раннему летальному исходу.
Тип учебного занятия: практическое занятие.
Цели занятия:
1. Учебная:
Сформировать представление о классификации генных болезней, механизмах их возникновения и патогенезе. Рассмотреть примеры наиболее распространенных и значимых генных заболеваний.
2. Развивающая:
Развитие у обучающихся углубленного понимания молекулярных основ генных болезней, их клинических проявлений и возможностей современной генетической диагностики и терапии.
3. Воспитательная:
Воспитательная цель занятия "Генные болезни" направлена на формирование у обучающихся глубокого понимания моральных, этических и социальных аспектов, связанных с генетическими заболеваниями. Это включает в себя развитие эмпатии к людям, страдающим от таких болезней, и осознание необходимости толерантного отношения к ним. Занятие призвано способствовать формированию научного мировоззрения, основанного на понимании молекулярных механизмов наследственности и генетических отклонений. Важно подчеркнуть роль генетических исследований в развитии медицины и возможности профилактики и лечения генных болезней. Кроме того, воспитательная цель заключается в формировании ответственного отношения к собственному здоровью и здоровью будущих поколений. Обучающиеся должны осознавать важность генетического консультирования и пренатальной диагностики для снижения риска рождения детей с наследственными заболеваниями.
Мотивация темы
Генные болезни – важная тема для специальности «Сестринское дело», поскольку медсестры играют ключевую роль в уходе за пациентами с такими заболеваниями и их семьями. Мотивация изучения этой темы обусловлена несколькими факторами.
Во-первых, понимание генетической основы заболеваний позволяет медсестрам лучше осознавать патофизиологические процессы, лежащие в основе симптомов, и, следовательно, оказывать более эффективную и персонализированную помощь.
Во-вторых, медсестры часто являются первым звеном, контактирующим с пациентами и их семьями, нуждающимися в генетическом консультировании. Знание основных принципов генетики позволяет им направлять пациентов к специалистам и предоставлять первичную информацию о заболеваниях.
В-третьих, медсестры участвуют в мониторинге эффективности лечения генных болезней, а также в выявлении и управлении побочными эффектами. Понимание генетических особенностей пациента помогает предвидеть реакцию на лекарственные препараты и корректировать план ухода.
Обучающийся (базовой подготовки) должен обладать общими и профессиональными компетенциями, включающими в себя способность:
ОК 01. Выбирать способы решения задач профессиональной деятельности применительно к различным контекстам;
ОК 02. Использовать современные средства поиска, анализа и интерпретации информации, информационные технологии для выполнения задач профессиональной деятельности;
ОК 03. Планировать и реализовывать собственное профессиональное и личностное развитие, предпринимательскую деятельность в профессиональной сфере, использовать знания по финансовой грамотности в различных жизненных ситуациях;
ПК 3.1. Консультировать население по вопросам профилактики заболеваний;
ПК 3.2. Пропагандировать здоровый образ жизни.
Данное практическое занятие способствует формированию у обучающихся следующих личностных результатов:
ЛР 7. Готовый соответствовать ожиданиям работодателей: проектно мыслящий, эффективно взаимодействующий с членами команды и сотрудничающий с другими людьми, осознанно выполняющий профессиональные требования, ответственный, пунктуальный, дисциплинированный, трудолюбивый, критически мыслящий, нацеленный на достижение поставленных целей; демонстрирующий профессиональную жизнестойкость;
ЛР 9. Принимающий активное участие в социально значимых мероприятиях, соблюдающий нормы правопорядка, следующий идеалам гражданского общества, обеспечения безопасности, прав и свобод граждан России; готовый оказать поддержку нуждающимся;
ЛР. 12. Способный искать нужные источники информации и данные, воспринимать, анализировать, запоминать и передавать информацию с использованием цифровых средств; предупреждающий собственное и чужое деструктивное поведение в сетевом пространстве;
ЛР. 13. Проявляющий навыки сотрудничества со сверстниками, детьми младшего возраста, взрослыми в образовательной, общественно полезной, учебно-исследовательской, проектной деятельности;
После изучения данной темы обучающийся должен:
уметь:
- определять аномальные фенотипы по фотографиям больных;
- выполнять тестовые задания;
- отвечать на вопросы.
знать:
- проявления генных болезней.
Междисциплинарные связи:
ОП. 01 Анатомия и физиология человека;
ОП. 02 Основы патологии;
ОП. 06 Фармакология;
МДК 03.01 Здоровый образ жизни и профилактика заболеваний в разные возрастные периоды;
МДК 04. 02 Сестринский уход и реабилитация пациентов терапевтического профиля разных возрастных групп.
Методы организации и осуществления учебно-познавательной деятельности:
- словесные
- наглядные
- практические
- диалогические
- объяснительно - иллюстративные
- проблемно-поисковые
Место проведения занятия: учебная аудитория ГБПОУ КК «Камчатский медицинский колледж».
Материально-техническое оснащение занятия:
- интерактивная доска;
- проектор;
- компьютер;
- презентация преподавателя;
- ситуационные задачи;
- тестовые задания.
Структурно-логическая схема практического занятия
| Название этапа занятия | Время | Действия преподавателя | Действия обучающегося |
| Организационный момент | 5 |
|
|
| Организационный момент
| 1 | Приветствует обучающихся, осуществляет контроль присутствия на занятии, готовности к занятию учебного кабинета и обучающихся, оценивает внешний вид обучающихся. | Занимают рабочие места, приветствуют преподавателя, слушают, отвечают. |
| Сообщение темы, ее обоснование | 1 | Сообщает тему, задает вопросы, побуждающие к обсуждению актуальности изучаемой темы. Организует обсуждение. | Записывают тему, участвуют в обсуждении. |
| Определение целей занятия, плана его проведения | 2 | Вовлекает обучающихся в постановку целей занятия: что должны знать, уметь; сообщает план работы на занятии. | Слушают, задают вопросы, участвуют в целеполагании. Записывают в тетрадях требования к знаниям, умениям по теме. |
| II. Контроль исходного уровня знаний | 10 |
|
|
| Фронтальный опрос
| 10 | Устный опрос (Приложение А) | Отвечают, задают уточняющие вопросы, участвуют в обсуждении. |
| III. Приобретение новых умений | 62 |
|
|
| Выполнение заданий по определению фенотипов | 20 | Раздает задания (приложение Б). Контролирует работу студентов | Выполняют задания, отвечают, оценивают ответы одногруппников. |
| Заполнение таблицы | 10 | Раздает таблицы (приложение В). Контролирует работу студентов | Заполняют таблицу |
| Решение ситуационных задач
| 32 | Проводит инструктаж, раздает задания (Приложение Г). Выслушивает ответы, проводит анализ ошибок.
| Решают устно задачи, отвечают, оценивают ответы одногруппников. |
| IV. Физкультминутка | 2 | Демонстрирует студентам технику выполнения упражнений (Приложение Д) | Выполняют совместно с преподавателем |
| V. Закрепление нового материала, контроль знаний, сформированности умений | 10 |
|
|
| Текущий срез знаний | 10 | Проводит инструктаж, раздает тестовые задания, проводит анализ ошибок. Озвучивает критерии оценивания (Приложение Е) | Выполняют тестовые задания. Совместно с преподавателем проводят анализ ошибок |
| VI. Подведение итогов занятия | 3 |
|
|
| Обобщение, подведение итогов занятия, оценка работы. Рефлексия. | 2 | Дает аргументированную оценку работы каждого обучающегося, подводит результаты достижения целей занятия, выставляет оценки. Раздает вопросы по рефлексии (Приложение Ж, Приложение З) | Осуществляют самооценку: соотносят результаты своей деятельности с целью занятия. |
| Домашнее задание | 1 | Проводит инструктаж по выполнению самостоятельной внеаудиторной работе (Приложение И) | Слушают, записывают в задание, задают вопросы. |
| Итого | 90 |
|
|
Список использованных источников
Основной источник
1. Бочков, Н. П. Медицинская генетика : учебник / под ред. Н. П. Бочкова. – Москва : ГЭОТАР-Медиа, 2016. - 224 с.
Методические пособия
1. Авилова, Т.М. Генетика человека. Наследственные болезни: Учебно-методическое пособие / Т.М. Авилова, Н.А. Мохаммад Амин, А.Н. Кривицкая. – Воглоград : Издательство ВолгГМУ, 2020. – 72 с.
2. Агаджанян, А.В. Медицинская генетика в иллюстрациях и таблицах : учебное пособие / А.В. Агаджанян, А.Ф. Фучич, Л.В. Цховребова, Р.И. Лазан-Турчич. - Москва : Практическая медицина, 2022. – 504 с.
3. Борисова, Т. Н. Генетика человека с основами медицинской генетики : учебное пособие для среднего профессионального образования / Т. Н. Борисова, Г. И. Чуваков. — 2-е изд., испр. и доп. — Москва : Издательство Юрайт, 2020. — 159 с.
4. Васильева, Е.Е. Генетика человека с основами медицинской генетики. Пособие по решению задач : учебное пособие для СПО / Е.Е. Васильева. – Санкт-Петербург : Лань, 2025. – 92 с.
5. Жилина, С.С. Генетика человека с основами медицинской генетики : учебник / С.С. Жилина, Т.В. Кожанова, М.Е. Майорова. – Москва : ГЭОТАР-Медиа, 2025. – 192 с.
6. Катмаков, П. С. Генетика : учебник для среднего профессионального образования / П. С. Катмаков, В. П. Гавриленко, А. В. Бушов, Е. И. Анисимова. — Москва : Издательство Юрайт, 2024. — 278 с.
7. Кургуз, Р.В. Генетика человека с основами медицинской генетики : учебное пособие для СПО / Р.В. Кургуз, Н.В. Кисилева. – Санкт-Петербург : Лань, 2024. – 176 с.
8. Иванищев, В.В. Основы генетики: учебник / В.В. Иванищев. – Москва : РИОР : ИНФРА – М, 2025. – 207 с.
9. Майорова, М.Е. Генетика человека с основами медицинской генетики : рабочая тетрадь / М.Е. Майорова. - Москва : ГЭОТАР-Медиа, 2024. – 64 с.
10. Рубан, Э.Д. Медицинская генетика : учебник / Э.Д. Рубан. – Ростов-на-Дону : Феникс, 2024. - 319 с.
Справочная литература:
1. Медицинская генетика : национальное руководство / под ред. Е.К. Гинтера, В.П. Пузырева, С.И. Куцева. - Москва : ГЭОТАР-Медиа, 2024. - 896 с.
2. Белецкая, Е.Я. Генетика и эволюция: справочник / Е.Я. Белецкая. – Москва : ФЛИНТА, 2020. – 108 с.
Приложение А
Контрольные вопросы для фронтального опроса:
1. Определение генных болезней
2. Классификация моногенных болезней
3. Особенности клинической картины генных болезней
4. Характеристика болезней аминокислотного обмена
5. Полные и мозаичные формы болезней
6. Прогредиентность клинической картины
7. Клинический полиморфизм
Эталоны ответов:
1. Генные болезни - разнородная по клиническим проявлениям группа заболеваний, обусловленных генными мутациями. Основой для объединения их в одну группу служат этиологическая генетическая характеристика и соответственно закономерности наследования в семьях и популяциях. Мутации в индивидуальных генах являются этиологическим фактором генных болезней поэтому закономерности их наследования соответствуют менделевским правилам расщепления в потомстве.
2. В основу классификации моногенных болезней положено несколько принципов:
По ведущей системной патологии: по органному и системному типу.
По этиологии. В этом случае выделяют 2 класса заболеваний: болезни с установленным первичным молекулярным (биохимическим) дефектом (10–11% всех МБ); болезни с неустановленным первичным молекулярным (биохимическим) дефектом. На эти заболевания приходится около 90% всех МБ.
По типу наследования патологического признака: аутосомно-доминантные; аутосомно-рецессивные; сцепленные с половой хромосомой доминантные и рецессивные; митохондриальные.
По преимущественному поражению того или иного вида обмена. Благодаря этому принципу многие МБ называются наследственными болезнями обмена веществ (НБО). Среди них выделено более 700 форм, в том числе 200 с установленным биохимическим дефектом. Среди НБО выделяют:
Болезни обмена аминокислот (фенилкетонурия, гомоцистинурия).
Болезни обмена углеводов (сахарный диабет, мукополисахаридозы).
Болезни обмена жиров (семейная гиперхолестеринемия).
Болезни пуринового и пиримидинового обмена (подагра, синдром Леша-Найхана).
Болезни соединительной ткани (синдром Марфана).
Болезни обмена ионов и минералов (муковисцидоз, гиперхроматоз).
Болезни обмена стероидов (адреногенитальный синдром).
Болезни обмена белков (миодистрофии, гемоглобинопатии: серповидно-клеточная анемия, талассемия).
3. К таким особенностям относятся многообразие проявлений, разный возраст начала болезни, прогредиентность клинической картины и хроническое течение, тяжесть течения, обусловливающая инвалидность с детства и меньшую продолжительность жизни.
4. Характерным почти для всех заболеваний этой группы является тип наследования — аутосомно-рецессивный, т. е. 25% потомства двух клинически здоровых носителей (гетерозигот) оказывается пораженным.
Общий биохимический признак — ацидоз тканей и аминоацидурия (согласно этому симптому называется вся группа заболеваний).
Неспецифические клинические признаки: рвота, обезвоживание организма (интоксикационный синдром), неврологические нарушения — летаргическое состояние или возбуждение, судорожный синдром. С возрастом появляется задержка психомоторного развития, регрессия приобретенных ранее навыков, умственная отсталость (вплоть до идиотии) и задержка физического развития.
5. Полные формы - формы, обусловленные гаметическими (в зародышевых клетках) мутациями. Это могут быть новые или унаследованные от предыдущих поколений мутации. В этих случаях патологические гены присутствуют во всех клетках организма. Однако любые мутации, в том числе и генные, могут возникать на ранних стадиях дробления зиготы в одной из клеток, и тогда индивид будет мозаичен по данному гену. В одних клетках у него будет функционировать нормальный аллель, в других - мутантный или патологический. Если эта мутация доминантная, то она проявится в соответствующих клетках и, очевидно, приведет к развитию менее тяжелой формы болезни. Если возникшая мутация в одной из клеток на ранних стадиях развития зародыша рецессивная, то ее эффект проявится только у гетерозиготы.
6. Течение большинства генных болезней тяжелое, что приводит к инвалидизации в детском возрасте и сокращению продолжительности жизни. При многих болезнях клиническая картина и тяжесть течения усиливаются по мере развития патологического процесса. Первичная биологическая основа этой характеристики - непрерывность функционирования патологического гена (либо отсутствие его продукта). К этому присоединяются вторичные процессы (воспаление, дистрофия, нарушенный обмен веществ, гиперплазия и т.д.), которые усиливают первично запущенный патологический процесс. Прогредиентность присуща не всем болезням.
7. Клинический полиморфизм генных болезней проявляется в разных сроках начала заболевания, полноте и тяжести симптоматики (глубина патологического процесса), продолжительности болезни, степени инвалидности, толерантности к терапии, в сокращении продолжительности жизни. Вместе с тем следует подчеркнуть, что генные болезни не имеют плавных переходов от нормы к патологии. Даже самая легкая форма болезни обязательно имеет минимальные диагностические критерии. Генетические причины клинического полиморфизма могут быть обусловлены не только патологическим геном, но и генотипом в целом, т.е. генотипической средой в виде генов-модификаторов. Вместе с патологическим геном индивид наследует от родителей комбинации других генов, которые могут усиливать или ослаблять действие патологического гена. В развитии генной болезни, как и любого наследственного признака человека, имеет значение не только генотип, но и внешняя среда.
Приложение Б
Задание по определению фенотипов
Используя представленный и лекционный материал, определите наследственные болезни по фотографиям пациентов.

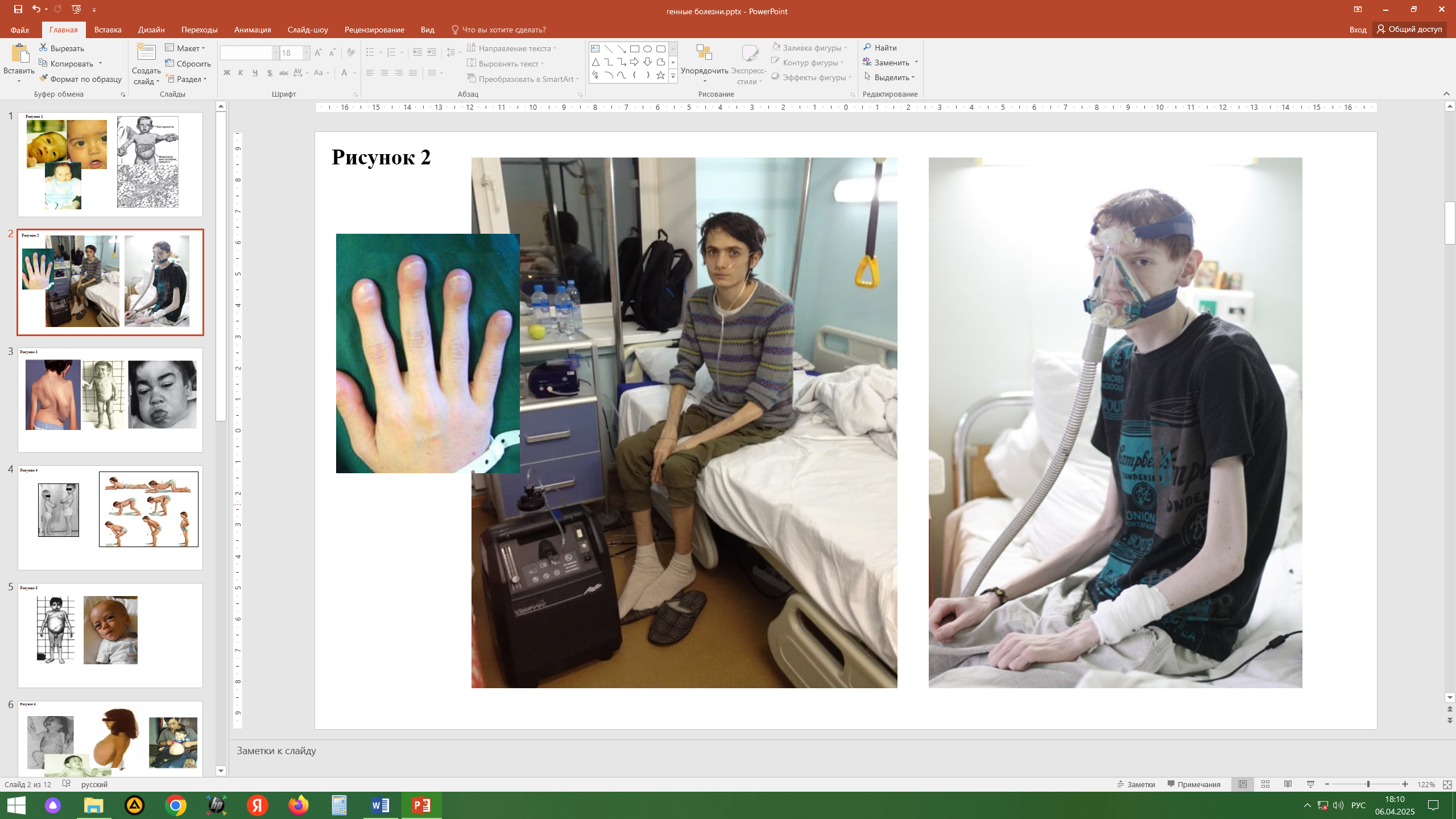
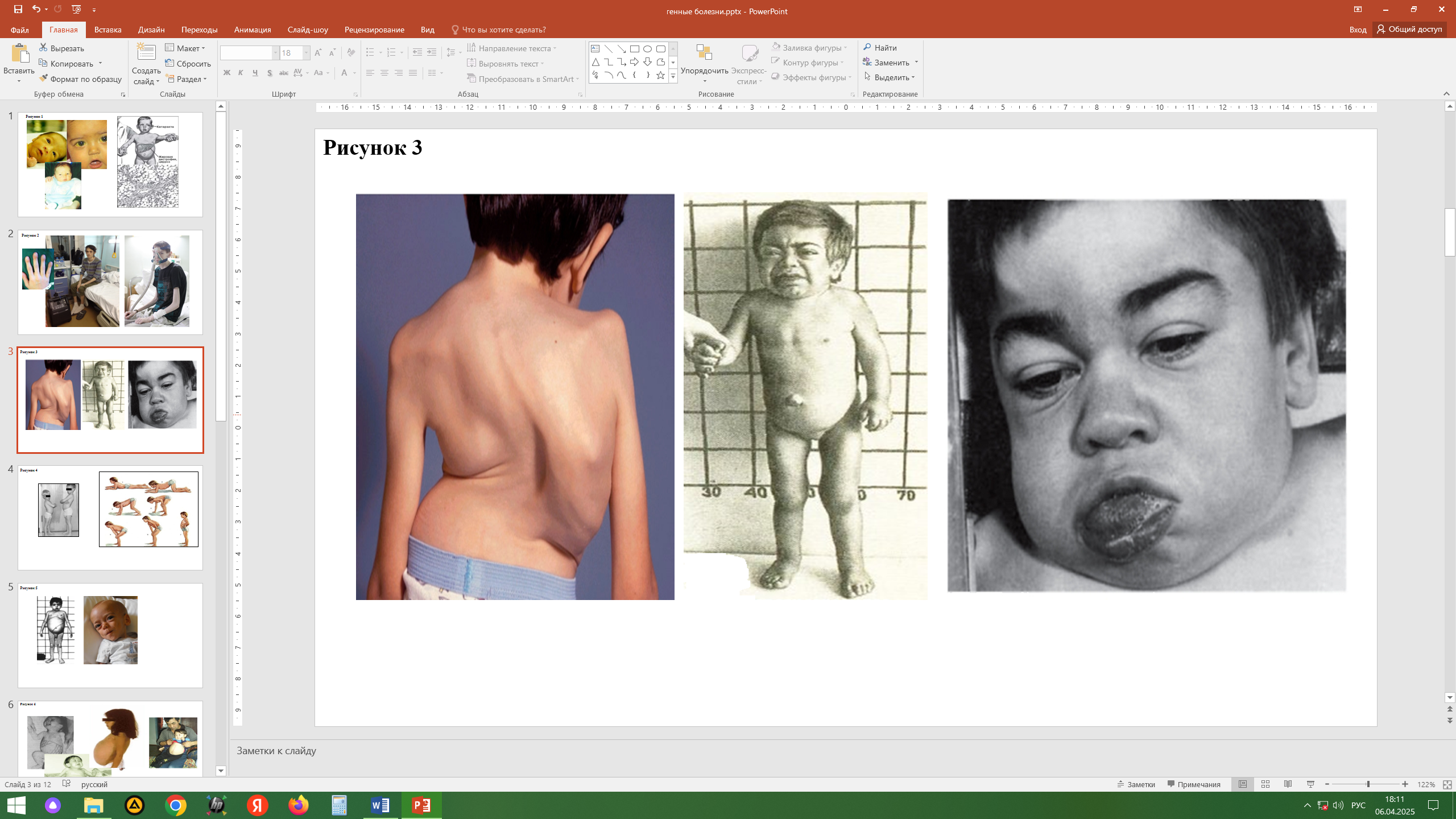






Синдром Марфана (болезнь соединительной ткани). Аутосомно-доминантный тип наследования: мутация в гене фибриллина (15 хромосома). Обнаружение связи гена фибриллина с синдромом Марфана дает возможность проводить молекулярно-генетическую диагностику, в том числе пренатальную. Частота синдрома Марфана в популяции равна 1 : 10 000-15 000. Характерно: нарушения мышечно-скелетной системы (арахнодактилия, высокий рост, длинные конечности, деформация позвоночника, ненормальная подвижность суставов), вывих хрусталика, сердечно-сосудистые изменения (аневризма аорты, пролапсы клапанов), вегетативно-сосудистые расстройства. Наиболее специфичными проявлениями для диагностики считаются вывих хрусталика, расширение аорты, расслоение аорты, эктазия твердой мозговой оболочки. При синдроме Марфана нередки врожденные пороки сердца, расстройства органов дыхания, поражения желудочно-кишечного тракта и мочевыводящей системы. Изредка может наблюдаться понижение слуха, глухота. Психическое и умственное развитие больных не отличается от нормы. Средняя продолжительность жизни составляет примерно 27 лет, хотя часть больных доживает до старости.
Муковисцидоз - наследственное заболевание с распространенным поражением экзокринных желез, кистозным перерождением поджелудочной железы, желез кишечника и дыхательных путей из-за закупорки их выводных протоков вязким секретом. Мутация в гене хромосомы 7, ответственного за синтез белка - муковисцидозного трансмембранного регулятора проводимости, обеспечивающего функцию хлоридного канала. Тип наследования - аутосомно-рецессивный. Нарушение синтеза белка - муковисцидозного трансмембранного регулятора проводимости приводит к избыточному выведению хлоридов и гиперсекреции густой слизи в клетках экзокринной части поджелудочной железы, эпителии бронхов, слизистой оболочки ЖКТ. Выделяют 4 формы заболевания: 1) мекониевый илеус новорожденных - избыточное заполнение кишечника густым меконием к моменту рождения (1%); 2) кишечная форма - недостаточность панкреатических ферментов, обильный зловонный стул, отставание в развитии (5-10%); 3) бронхолегочная форма - вязкий секрет приводит к обструктивному синдрому и присоединению вторичной инфекции (15-20%); 4) смешанная форма (65-75%). Диагностика муковисцидоза основана на клинической картине, результатах биохимического определения ионов натрия и хлора в поте. Основными направлениями лечения больных с муковисцидозом являются: назначение микросферических панкреатических ферментов с рН-чувствительной оболочкой, антибиотикотерапия (раннее начало, длительное лечение, применение с профилактической целью), прием урсодезоксихолевой кислоты. Также в настоящее время активно ведутся разработки в области генной терапии заболевания и поиск веществ, способных стимулировать синтез, транспорт и функции неполноценного кистофиброзного трансмембранного регулятора
Синдром адреногенитальный - врожденное патологическое состояние, обусловленное дисфункцией коры надпочечников с чрезмерной секрецией андрогенов и проявляющееся признаками вирилизации. Тип наследования - аутосомно-рецессивный. Частота в популяции 1:800010000. В патогенезе имеет место недостаточность фермента 21-гидроксилазы - 95% случаев. Известны два классических варианта этой болезни - сольтеряющая и простая вирильная формы. Сольтеряющая форма адреногенитального синдрома – полный дефицит фермента, с нарушением солевого обмена (дефицит минералокортикоидов). У новорожденного появляются срыгивание, рвота, симптомы недостаточности периферического кровообращения, сонливость, потеря массы тела, обезвоживание. Простая вирильная форма - ускоренное соматическое развитие, повышение экскреции гормонов коры надпочечников. У девочек при нормальном кариотипе мускулинизация различной выраженности. Вирильная форма проявляется избытком андрогенов: у девочек - врожденные изменения гениталий, мышечная масса по мужскому типу, грубый голос. У мальчиков - макрогенитосомия, преждевременное половое созревание (к 5-7 годам). Неклассические и поздние формы заболевания клинически проявляются только в подростковом возрасте. Поздняя диагностика, а также несвоевременное и некорректное лечение приводят к тяжелым последствиям: гибели ребенка от сольтеряющих кризов, ошибкам в выборе половой принадлежности при выраженной вирилизации гениталий у девочки, нарушениям роста и полового созревания, а также к бесплодию. Эффективной оказывается терапия глюкокортикоидами и минералокортикоидами, хотя не все проявления заболевания поддаются коррекции.
Миодистрофия Дюшенна - тяжелое наследственное заболевание с повышенной активностью в плазме крови ряда мышечных ферментов, особенно креатинкиназы. Встречается с частотой 1:3500 новорожденных мальчиков. Наследование сцеплено с полом, рецессивное. Дети с дистрофией Дюшенна начинают поздно ходить, походка изменена. Характерны атрофия мышц, атрофический процесс в сердечной мышце, нарушение моторики кишечника, изменение в костной системе, снижение интеллекта. На самой последней стадии атрофия (слабость) захватывает мышцы лица, глотки и дыхательные мышцы. Продолжительность жизни 20-30 лет. Из биохимических показателей для миодистрофии Дюшенна наиболее характерен резко повышенный (в 10-100 раз) уровень креатинфосфокиназы в сыворотке крови. В настоящее время разрабатывается терапия миодистрофии: генная (заместительная), клеточная (миобласты, стволовые клетки), аминогликозидная терапия, применение ингибиторов протеосом и др.
Фенилкетонурия
Частота 1:10 000. Ребенок рождается здоровым. Фенотипические признаки — светлые волосы, светлая кожа, голубые глаза. Генный деффект приводит к дефициту фермента фенилаланингидроксилазы и препятствует превращению фенилаланина в тирозин (ген локализован в длинном плече 12-й хромосомы). Концентрация фенилаланина в крови повышается, и он включается в побочный метаболический цикл (трансаминирование), превращаясь в токсичные для развивающейся нервной системы ребенка фенилпировиноградную, фенилуксусную кислоты и др. фенилкетоновые производные. Главным следствием токсического воздействия является умственная отсталость (в 65% - глубокая). Фенилпировиноградная кислота выделяется с мочой, придавая ей особый «мышиный» запах. Дефицит тирозина сказывается на образовании пигмента меланина, в меньшей степени страдает обмен тиреоидных гормонов и катехоламинов. В связи с этим у больных с ФКУ кожа депигментирована, обладает повышенной чувствительностью к ультрафиолету, часто развиваются экземы и дерматиты, волосы светлые и светлый цвет радужной оболочки глаза. Характерна специфическая поза: локтевые, тазобедренные и коленные суставы слегка согнуты, туловище наклонено вперед. Начало диетолечения в первые недели жизни и на протяжении 10-12 лет позволяет в 90% случаев предупредить развитие умственной отсталости. Клинические симптомы ФКУ (умственная отсталость, судорожный синдром, гиперкинезы, походка, поза «портного», склонность к дерматитам) проявляются через 3–6 месяцев после рождения. Основной биохимический маркер ФКУ — увеличение плазменной концентрации фенилаланина — определяется через 3–4 дня после начала кормления. Биохимические диагностические критерии: проба Феллинга (скрининг-тест): моча зеленого цвета; тест Гатри; повышенный уровень в моче метаболитов фенилаланина, «мышиный» запах мочи; снижение толерантности к полученному внутрь фенилаланину. Вовремя начатое лечение (диетотерапия) обеспечивает хороший клинический эффект, нормальную продолжительность жизни.
Альбинизм
Распространенный наследственный дефект пигментации, часто обусловленный дефектом синтеза фермента тирозиназы, необходимой для нормального меланинообразования. Частота 1:39 000. Тотальная депигментация кожи, волос, глаз, причем окраска одинакова для всех расовых групп и не меняется с возрастом. Кожа не загорает, совершенно отсутствуют невусы, какие-либо пигментные пятна. Острота зрения значительно снижена и с возрастом не улучшается. Снижена резистентность к инфекциям. Возможны эпилепсия, бесплодие. Предрасположенность к раку кожи. Традиционные методы лечение альбинизма неэффективны. Рекомендуется использовать различные средства защиты от ультрафиолетовых лучей.
Болезнь Нимана — Пика (сфингомиелиновый липидоз)
Частота 1:100 000. Происходит накопление липида сфингомиелина и вторично миелина в клетках нервной ткани (преимущественно в головном мозге) и паренхиматозных органах (печени,
селезенке). Клиника: болезнь проявляется в 4–6 месяцев; вторичная гипотрофия вследствие повторных рвот и отказа от приема пищи; кожа кофейно-желтой окраски; отставание
в нервно-психическом развитии; глухота, слепота; снижение резистентности к инфекционным заболеваниям; летальный исход к 3 годам.
Наследование аутосомно-рецессивное. Диагностика — анализ крови на фермент. Лечение — поддерживающая терапия.
Болезнь Тея-Сакса (ранняя детская идиотия амовратическая) - аутосомно-рецессивное заболевание, частота встречаемости 1:25000, ген картирован на хромосоме 15. Мутация в гене, ответственном за синтез фермента гексозоаминидазы А, принимающего участие в метаболизме ганглиозидов. В возрасте около полугода возникает регресс в психическом и физическом развитии. Ребенок теряет зрение, слух, способность глотать, появляются судороги. Характерным симптомом является наличие красного пятна на сетчатке напротив зрачка (симптом «вишневой косточки»). Постепенно мышцы атрофируются и наступает паралич, продолжительность жизни до 4 лет.
Галактоземия
Частота 1:35 000–50 000. Нарушение процесса ферментативного превращения галактозы в глюкозу с накоплением галактозы и продуктов ее обмена в клетках, что оказывает повреждающее действие на функции печени, нервной системы и другие органы. Клиника: желудочно-кишечные расстройства — понос и рвота с первых дней жизни ребенка (после чайно-водной паузы состояние улучшается, но введение молока обуславливает рецидив нарушений со стороны ЖКТ); проявления недостаточности печени: стойкая желтуха с преобладанием в крови прямого билирубина, увеличение печени, цирроз печени; помутнение хрусталика (катаракта появляется позднее); задержка психофизического развития; поражение почек, увеличение селезенки, асцит, судороги, нарушение сосания, глотания, мышечная гипотония. Характерна задержка психического развития и умственная отсталость. Наследуется по аутосомно-рецессивному типу. Диагностика: 1) определение концентрации галактозо-1-фосфата в эритроцитах (повышена); 2) повышение уровня галактозы в крови и моче; 3) микробиологический тест Гатри.
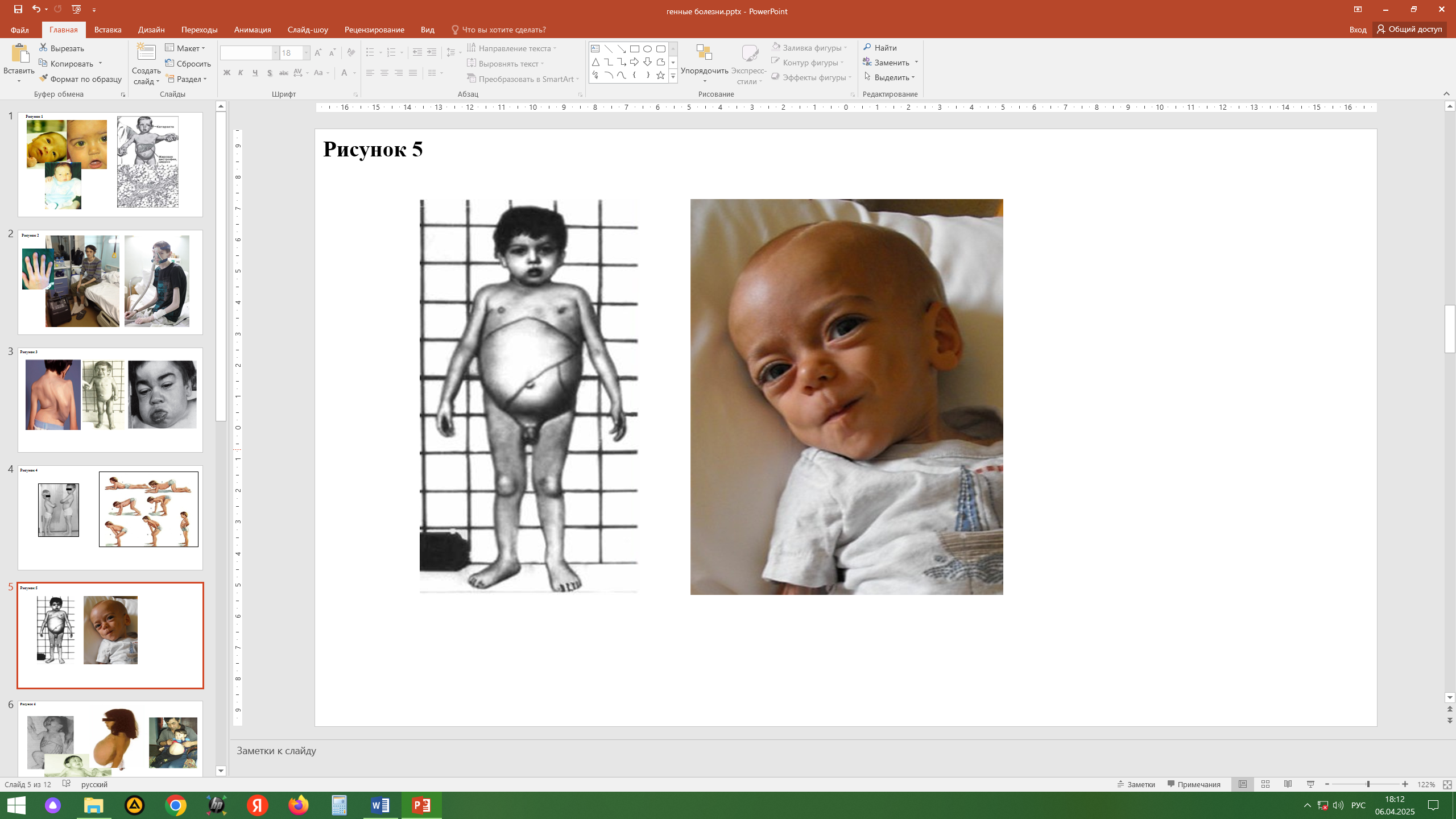
Мукополисахаридозы (МПС)
Гетерогенная группа заболеваний, отнесенных к наследственным болезням обмена сложных сахаров. МПС сопровождаются избыточным накоплением в тканях и повышенной экскреции гликозаминогликанов (ГАГ) — кислых мукополисахаридов, соединенных с белком и состоящих из уроновых кислот, аминосахароз и нейтральных сахаров. Указанные комплексы существуют в форме протеогликанов, являющихся важнейшими компонентами основного структурного белка волос (α-кератин) и структурного белка соединительной ткани (коллаген). Тип наследования - аутосомно-рецессивный и рецессивный Х-сцепленный. Клиническая картина: манифестация болезни, как правило, в возрасте до 7 лет, задержка роста (до карликовости), кифоз/кифосколиоз/сколиоз, массивный череп с глубоким и удлиненным турецким седлом, короткая шея, деформация грудной клетки, веслообразные ребра, укорочение трубчатых костей, грубые черты лица, помутнение роговицы, задержка психического развития, судороги, глухота, врожденные пороки сердца. Характерные для МПС признаки дисморфизма получили название «гаргоилический фенотип» (гаргоилизм — синоним МПС). Диагностика МПС основывается на совокупности данных генеалогического анализа, клинических проявлений, типичных рентгенологических данных, экскреции с мочой оксипролина (снижение), ГАГ и их фракций (превышение в 5–10 раз). Выделено 9 патогенетически различных заболеваний, но все они - результат нарушения обмена кислых мукополисахаридов, играющих важную роль в функционировании соединительной ткани. Лечение больных МПС в основном симптоматическое, заключается в назначении терапии, способствующей нормализации или стабилизации патологического процесса в опорно-двигательном аппарате, сердечно сосудистой и цен тральной нервной системе, паренхиматозных органах, органах зрения и слуха. Перспективным считается плазмофорез.
Эталоны ответов:
1. галактоземия
2. муковисцидоз
3. мукополисахаридоз
4. миодистрофия Дюшенна
5. болезнь Ниманна-Пика
6. альбинизм
7. болезнь Тея-Сакса
8. синдром Марфана
9. фенилкетонурия
10. адреногенитальный синдром
Приложение Б
Таблица «Сравнительная характеристика генных болезней»
Задание: Заполните таблицу, используя материал учебника.
| Болезнь | Частота встречаемости | Тип наследования, причина | Проявления заболевания | Методы диагностики | Лечение |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Приложение В
Ситуационные задачи
Критерии оценивания:
- оценка «отлично»: ответ на вопрос задачи дан правильно. Объяснение хода ее решения подробное, последовательное, грамотное, с теоретическими обоснованиями (в т.ч. из лекционного курса), с необходимым схематическими изображениями и демонстрациями алгоритмов, с правильным и свободным владением медицинской терминологией; ответы на дополнительные вопросы верные, четкие.
- оценка «хорошо»: ответ на вопрос задачи дан правильно. Объяснение хода ее решения подробное, но недостаточно логичное, с единичными ошибками в деталях, некоторыми затруднениями в теоретическом обосновании (в т.ч. из лекционного материала), с необходимым схематическими изображениями и демонстрациями алгоритмов, с правильным и свободным владением медицинской терминологией; ответы на дополнительные вопросы верные, но недостаточно четкие.
- оценка «удовлетворительно»: ответ на вопрос задачи дан правильно. Объяснение хода ее решения недостаточно полное, непоследовательное, с ошибками, слабым теоретическим обоснованием (в т.ч. лекционным материалом), со значительными затруднениями и ошибками в схематических изображениях и алгоритмах, ответы на дополнительные вопросы недостаточно четкие, с ошибками в деталях.
- оценка «неудовлетворительно»: ответ на вопрос дан неправильно. Объяснение хода ее решения дано неполное, непоследовательное, с грубыми ошибками, без теоретического обоснования, и демонстраций алгоритмов или с большим количеством ошибок, ответы на дополнительные вопросы неправильные (отсутствуют).
Задача 1. У ребенка 5 лет нарушен тирозиновый обмен. Положительная реакция пробы Феллинга, затхлый специфический запах мочи и пота, замедленное моторное и психическое развитие с 6-месячного возраста. Это заболевание легко лечится специальной диетой, назначенной в раннем возрасте. О каком заболевании может идти речь у больного?
Задача 2. У ребенка при грудном вскармливании появились пожелтения кожи, увеличение печени. Проба с хлористым железом отрицательная. Врач назначил вместо грудного молока специальную диету, это улучшило состояние ребенка. Какое заболевание у этого ребенка?
Задача 3. К врачу обратился больной с жалобами на непереносимость солнечной радиации. Имеются ожоги кожи и нарушение зрения. Нарушение обмена какой аминокислоты отмечается у этого пациента?
Задача 4. В больницу обратились супруги с 9-месячным ребенком с гипотрофией, но психически нормально развитым. Ребенок болеет почти с периода новорожденности, страдает коклюшеподобным кашлем. С 5 месяцев, после введения прикорма, появились частые дефекации с большим количеством светлых каловых масс с неприятным запахом. Отмечается увеличение печени. По лабораторным исследованиям — повышение концентрации натрия и хлора в поту. Какое заболевание можно предположить?
Задача 5. Мальчик 8 мес. Родители обратились к врачу с жалобами на отставание ребенка в психомоторном развитии. Из анамнеза известно, что ребенок родился в срок от нормально протекавшей беременности, с массой тела 3500 г. Роды нормальные, период новорожденности протекал без особенностей. Первые месяцы жизни развивался нормально: следил за предметами, улыбался, хорошо удерживал голову, опирался на ножки, лежа на животе опирался на руки. В 4-4,5 мес. было замечено, что ребенок стал вздрагивать при внезапном звуке, перестал улыбаться, интересоваться игрушками, стал безразличным. На протяжении последующих трех месяцев утратил приобретенные ранее двигательные навыки. При осмотре в возрасте 8 мес.: телосложение правильное, удовлетворительного питания. Взгляд на предметах не фиксирует. Создается впечатление, что ребенок не видит. Самостоятельно не сидит, на ноги не опирается, движения в конечностях не координированные, размашистые. Мышечный тонус в конечностях равномерно снижен. Сухожильные рефлексы высокие, брюшные отсутствуют. Нарушений чувствительности нет. Ваш предварительный диагноз?
Эталоны ответов
1. Фенилкетонурия
2. Галактоземия
3. Альбинизм
4. Муковисцидоз
5. Болезнь Тея-Сакса
Задачи для одаренных обучающихся
Задача 1. Мальчик 6 мес. направлен на консультацию к кардиологу. Ребенок от 1-й беременности (с ОРВИ на 10-й неделе, угрозой выкидыша), срочных родов, с массой 3100 г, длиной 52 см, закричал сразу. Грудь сосал вяло, ежемесячная прибавка массы тела на первом году - 400-500 г. Вскармливание естественное. Трижды перенес ОРВИ. Анамнез: у мамы - хронический тонзиллит, у брата 8 лет - ВПС, у бабушки по линии матери - ИБС, по линии отца: у сестры - ревмокардит, дедушка умер от рака желудка, у бабушки - ГБ. Осмотр: состояние ребенка средней степени тяжести, беспокойный, кожа бледная, акроцианоз. Ушные раковины имеют аномальную форму. Арахнодактилия. ЧД 40 в мин., дыхание пуэрильное с единичными влажными хрипами, при перкуссии легочный звук. Верхушечный толчок в 4-5 межреберье, усиленный. Тоны сердца громкие, систолический шум во всех точках аускультации с эпицентром во 2 м/р слева от грудины, проводится на спину. Живот мягкий, безболезненный, печень выступает из-под края реберной дуги на 2,5 см. Мочеиспускание свободное. Поставьте предварительный диагноз.
Задача 2. В инфекционную больницу поступила девочка 2-х месяцев. Со слов матери с пятого дня жизни у ребенка отмечается рвота, жидкий стул после каждого кормления, девочка находится на естественном вскармливании. В 1 месяц появилось желтушное окрашивание кожи. Осмотр: ребенок пониженного питания, масса тела ниже возрастной нормы. Кожный покров желтушный, печень выступает из-под края реберной дуги на 5 см. Двигательная активность понижена, не фиксирует взгляд, не улыбается. Из анамнеза известно, что что старший брат девочки умер в возрасте 5 лет от печеночной недостаточности.
Задача 3. В больницу поступил ребенок 5 лет с пупочной невправляемой грыжей. Осмотр: массивный череп, короткая шея, грубые черты лица, грудная клетка деформирована, веслообразные ребра, роговица помутневшая, задержка нервно-психического развития. При пальпации живота в области пупка обнаруживается выпячивание не вправляемое в брюшную полость, отмечается увеличение печени и селезенки. В анализе мочи гликозаминогликонурия.
Задача 4. Девочка, 5 лет, поступила в клинику с жалобами на затрудненное дыхание, мучительный кашель, резкое истощение. Анамнез: больна с раннего возраста, анорексия, задержка физического развития, частые простудные заболевания. Психическое развитие соответствует возрасту. Мать и отец здоровы, дедушка по линии матери страдает хроническим бронхитом, у бабушки по линии отца - хронический колит, послеоперационная грыжа. Осмотр: состояние тяжелое, температура тела 390С, бледно-серая кожа. Мучительный кашель, мокрота слизисто-гнойного характера, отделяется с трудом. Рост 125 см, масса 16 кг. Грудная клетка деформирована. Перкуторный звук с коробочным оттенком, местами - с притуплением; аускультативно: дыхание проводится неравномерно, рассеянные влажные разнокалиберные хрипы. Тоны сердца приглушены. Живот увеличен в размерах, пальпируются вздутые кишечные петли. Стул нерегулярный, обильный, зловонный, с жирным блеском.
Задача 5. Ребенок 2,5 лет, от 2-й беременности (с токсикозом 1-й половины, нефропатией во 2-й половине), срочных родов, с массой 3400 г, длиной 52 см. Период новорожденности протекал удовлетворительно. Сидит с 7 месяцев, стоит с 11 месяцев, ходит с 2-х лет. Семейный анамнез: родители мальчика здоровы, матери 23 года, отцу 28 лет, первый ребенок здоров. С 5,5 месяцев: уплощение и облысение затылка, размягчение краев большого родничка. В возрасте 1 года 9 месяцев появились варусные деформации костей, «утиная походка». Осмотр: задержка физического развития, варусные деформации нижних конечностей, мышечная гипотония, кариес зубов. В анализе крови: увеличена активность щелочной фосфатазы, снижено содержание кальция и фосфора. В анализе мочи: снижено содержание кальция, фосфора. Рентгенография трубчатых костей: системный остеопороз, метафизы расширены, контуры неровные; расслоение надкостничного слоя.
Эталоны ответов
1. Синдром Марфана.
2. Галактоземия.
3. Мукополисахаридоз.
4. Муковисцидоз, смешанная форма.
5. Витамин D-зависимый рахит.
Приложение Г
Физкультминутка
1. “Пальцы в замке”
Простая пальчиковая гимнастика предназначена для расслабления мышц пишущей руки. Соедините руки и скрестите пальцы в замок, а затем выполняйте круговые движения в отдельности каждым пальцем. Соседние пальцы должны оставаться неподвижными.
2. “Ухо – нос”
Это упражнение помогает улучшить память, внимание и концентрацию. Одну ладонь поставьте на нос, а вторую, перекрестив с левой, – поднесите к уху. Хлопните в ладоши и поменяйте руки местами. Повторите упражнение несколько раз.
3. “Победитель”
Разнотипные движения рук развивают межполушарное взаимодействие, а также синхронизируют работу полушарий головного мозга. Одной ладонью покажите знак “Ок”, второй – “V”. Чередуйте руки.
4. “Невидимые картинки”
Цель упражнения – развитие вербальной памяти, внимания. Один обучающийся должен нарисовать картинку в воздухе, а другой – угадать, что он изобразил. Затем ребята меняются ролями. Изображать можно все, что угодно: еду, места, людей, животных.
5. “Вдох–выдох”
Выключите свет и включите тихую, расслабляющую музыку. Учащиеся закрывают глаза, делают несколько глубоких вдохов. Такая произвольная медитация очищает разум и позволяет мозгу осмыслить полученную информацию.
Приложение Е
Текущий срез знаний
Критерии оценивания:
- оценка 5 «отлично» выставляется за правильные ответы на 91-100 процентов заданий (9 и более правильных ответов);
- оценка 4 «хорошо» за правильные ответы на 80-89 процентов заданий (8 правильных ответов);
- оценка 3 «удовлетворительно» за правильные ответы на 70-79 процентов заданий (7 правильных ответов);
- оценка 2 «неудовлетворительно» за правильные ответы на 69 процентов заданий и менее (менее 7 правильных ответов).
Вариант 1
Выбрать один правильный ответ
1. Признак наследственной патологии
1) вовлеченность в патологический процесс нескольких органов и систем
2) манифестация, строго определенная во времени
3) вовлеченность в патологический процесс всех членов семьи
4) волнообразное течение
2. Синдром Марфана связан с аномалиями
1) костной системы, что приводит к увеличению длины и толщины костей
2) мышечной ткани, что приводит к слабости сухожилий и связок
3) соединительной ткани (дилатация аорты и разболтанность суставов)
4) гормона роста (избыточное отложение жира, высокий рост)
3. Для нейрофиброматоза характерно
1) заболевание выявляется у одного из родителей пробанда
2) экспрессивность гена в одной семье
3) наличие множественных пигментных невусов
4) все вышеперечисленное
4. Критерии диагностики миодистрофии
1) прогрессирующая мышечная слабость
2) псевдогипертрофия икроножных мышц
3) повышение креатинфосфокиназы в сыворотке крови
4) все перечисленные
5. Диагностические критерии адреногенитального синдрома
1) гипертелоризм, брахидактилия, крипторхизм, низкий рост
2) гонады представлены яичками, гипогонадизм
3) прогрессирующая вирилизация, ускоренное соматическое развитие,
повышенная экскреция гормонов коры надпочечников
4) умственная отсталость, макроорхизм, оттопыренные уши
6. Грубые черты лица, гепатомегалия и умственная отсталость отмечается при
1) мукополисахаридозе
2) нарушение углеводного обмена
3) аминоацидурии
4) недостаточности ферментов цикла мочевины
6. Диагностические критерии муковисцидоза
1) хронические бронхоэктазы, правостороннее расположение сердца
2) грубые черты лица, кифосколиоз, порок клапанов сердца
3) рецидивирующие хронические пневмонии, нарушение функции
поджелудочной железы, мальабсорбция, обильный зловонный стул
4) задержка роста, помутнение роговицы
7. При болезни Тея-Сакса, которая наследуется аутосомно-рецессивно, развиваются необратимые изменения центральной нервной системы, приводящие к смерти в раннем детском возрасте. С нарушением обмена каких веществ связан этот наследственный дефект?
1) минеральных веществ
2) липидов
3) углеводов
4) аминокислот
8. У новорожденного наблюдается желтуха, рвота и понос. При обследовании было отмечено увеличение печени и селезенки, при лабораторном исследовании — галактоза и белок в моче. Каков наиболее вероятный диагноз?
1) тирозиноз
2) синдром Дауна
3) фенилкетонурия
4) галактоземия
9. У ребенка молочно-белая кожа, очень светлые волосы, светло-серые глаза, красные зрачки. Имеют место ожоги кожи при воздействии солнечного света и нарушение зрения. Предварительный диагноз — альбинизм. Обмен какой аминокислоты нарушен?
1) пролина
2) тирозина
3) аланина
4) триптофана
10. Наследственная болезнь, поддающаяся коррекции диетой
1) нейрофиброматоз
2) фенилкетонурия
3) муковисцидоз
4) умственная отсталость с ломкой Х-хромосомой
Вариант 2
Выбрать один правильный ответ
1. Для фенилкетонурии характерно
1) фенилкетонурия аутосомно-рецессивное заболевание
2) одним из признаков фенилкетонурии является гипопигментация
3) назначение диеты позволяет избежать задержки умственного развития
4) все перечисленные признаки
2. Адреногенитальный синдром возникает при нарушении обмена
1) белков
2) жиров
3) стероидов
4) углеводов
3. Диагностические критерии нейрофиброматоза
1) врожденный порок сердца и лучевой кости и ее производных
2) анемия, гепатоспленомегалия, «башенный» череп
3) себорейные аденомы на щеках, депигментированные пятна
4) множественные пигментные пятна на коже; опухоли кожные,
подкожные и по ходу нервных волокон, сколиоз, глиомы зрительного
нерва
4. Для нарушения обмена аминокислот характерно
1) метаболический ацидоз в раннем возрасте
2) необычный запах мочи и пота
3) сонливость, заторможенность, рвота у ребенка раннего возраста
4) все перечисленные признаки
5. Для наследственной патологии характерно
1) полиорганность поражения, резистентность к терапии
2) острое течение
3) благоприятный исход заболевания
4) отсутствие хронизации процесса
6. Проба Феллинга помогает диагностировать
1) муковисцидоз
2) гипотиреоз
3) фенилкетонурию
4) миодистрофию
7. У ребенка наблюдается слабая пигментация кожи, светлые волосы, голубые глаза. Родители — брюнеты. Внешне при рождении ребенок выглядел нормально, но на протяжении последних трех месяцев наблюдается повышенная возбудимость и тонус мышц, тремор, судорожные припадки. Причиной такого состояния может быть:
1) фенилкетонурия
2) галактоземия
3) гликогеноз
4) острая порфирия
8. У грудного ребенка с задержкой развития, мышечным гипертонусом и склонностью к самоповреждениям обнаружено повышенное содержание мочевой кислоты в крови. При каком метаболическом нарушении это наблюдается?
1) синдроме Леша — Нихана
2) подагре
3) синдроме приобретенного иммунодефицита
4) болезни Вильсона-Коновалова;
9. Укажите тип наследования галактоземии
1) сцепленное с У-хромосомой
2) сцепленный с Х-хромосомой
3) аутосомно-доминантный
4) аутосомно – рецессивный
10. Для диагностики моногенных заболеваний используются методы:
1) исследование полового хроматина
2) биохимический
3) клинико-генеалогический метод
4) цитогенетический
Эталоны ответов
| Вариант 1 | Вариант 2 |
| 1. 1 | 1. 4 |
| 2. 3 | 2. 3 |
| 3. 4 | 3. 4 |
| 4. 4 | 4. 4 |
| 5. 3 | 5. 1 |
| 6. 3 | 6. 3 |
| 7. 2 | 7. 1 |
| 8. 4 | 8. 1 |
| 9. 2 | 9. 4 |
| 10. 2 | 10. 2 |
Приложение Ж
Рефлексия
Карточки с заданием «Продолжи предложение», каждый студент отвечает на 1 карточку.
| Продолжи одно любое предложение Я получил(а) важные знания по……… или Я не узнал(а) для себя ничего нового…………., так как……… |
| Для меня сегодня остался невыясненным вопрос по………………………………….. (либо такового нет) |
|
Самым трудным для меня сегодня было…………., поэтому я……………………
|
| Продолжи одно любое предложение Сегодня мне было интересно………………………….. или Сегодня мне не понравилось …………………………и для этого мне нужно… |
|
Я считаю, что данная тема в дальнейшей профессиональной деятельности необходима для ………………………………………..
|
|
Если бы я вела данный урок, то я бы ………………………….. |
Приложение З
Оценочный лист
| № п/п | Фамилия, имя студента | Фронтальный опрос | Определение болезней по фотографиям | Заполнение таблицы | Решение ситуационных задач | Текущий срез знаний | Итоговая оценка |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Приложение И
Внеаудиторная самостоятельная работа
Виды работ:
1. Бочков, Н. П. Медицинская генетика : учебник / под ред. Н. П. Бочкова. – Москва : ГЭОТАР-Медиа, 2016. – С. 115-149
2. Подготовить сообщение и презентацию (темы: «Нарушение обмена аминокислот, углеводов и липидов», «Наследственные синдромы нарушения всасывания», «Распространенные психические и нервные болезни», «Распространенные болезни среднего возраста»).*
3. Заполнить таблицу «Генные болезни».*
| Генные болезни | Примеры |
| Заболевания, связанные с нарушением обмена углеводов |
|
| Заболевания, связанные с расстройством аминокислотного обмена |
|
| Заболевания, связанные с нарушением липидного обмена |
|
| Заболевания, связанные с нарушением обмена азотистых оснований |
|
| Заболевания, связанные с нарушением обмена металлов |
|
| Заболевания порфиринового и билирубинового обмена |
|
* - задание для одаренных обучающихся
25









 "Генные болезни" (15 MB)
"Генные болезни" (15 MB)
 0
0 142
142 0
0 Нравится
0
Нравится
0


